Variant Classifier
Overview
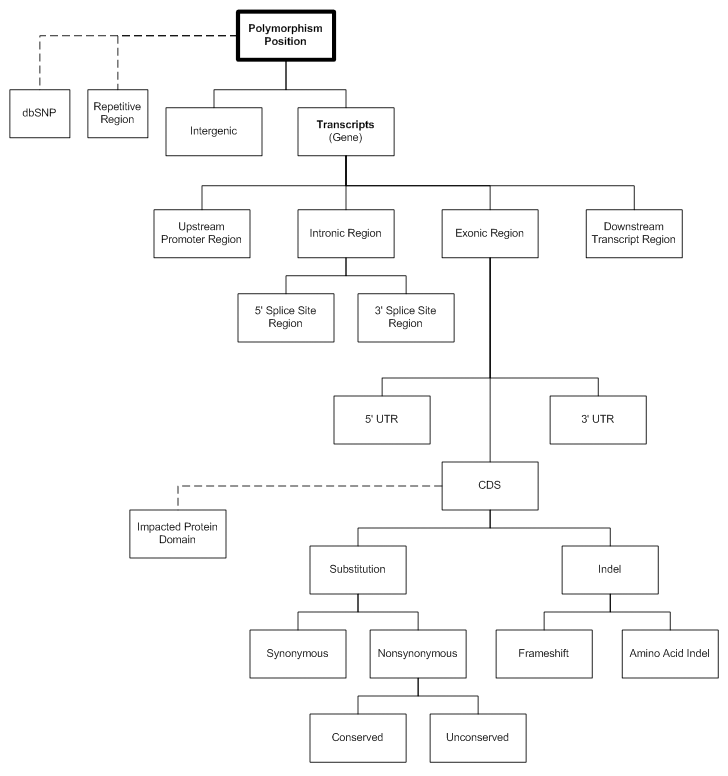
The VariantClassifier is a software tool for hierarchically classifying variants based on the genome annotation that is provided. Instead of looking at a region of the genome and seeing all the features relative to each other on the genomic axis, the VariantClassifier inverts the process so that novel variants can be tested for interest, based on the known features on the genomic axis. Furthermore, our hierarchical classification provides a prioritization of the variants that should be considered for more intensive study.
Depending on the depth of annotation information available, the VariantClassifier may assign your variant to a location such as: intergenic region, promoter region, intron, exon. If the variant is found near or on an exon the following may applicable and assigned as well: 3' splice site, 5' splice site, 5' UTR, CDS, or 3' UTR. If the variant is found in the CDS, then synonymous, non-synonymous, conserved, unconserved, frameshift, and indel may be applicable and assigned to your variant. If repetitive regions or known SNP coordinates are in the annotation, the VariantClassifier will report overlap with these features as well.
Classification Hierarchy Diagram